
 https://orcid.org/0000-0002-5408-6263
https://orcid.org/0000-0002-5408-6263
Las crisis convulsivas son descargas eléctricas anormales que tienen manifestaciones clínicas variadas, de origen multifactorial, y que se asocian a trastornos clínicos1.
Una convulsión es una contracción involuntaria de la musculatura, una alteración paroxística de la actividad motora y/o de la conducta limitada en el tiempo tras una actividad eléctrica anormal en el cerebro que puede deberse a mecanismos diferentes como por ejemplo: anóxico, metabólico, epiléptico y febril. Las crisis comiciales son frecuentes en el grupo de edad pediátrica y aparecen en un 10% de los niños. La mayor parte de las crisis en los niños se deben a trastornos somáticos que se originan fuera del cerebro, tales como fiebre elevada, infección, síncope, traumatismo craneal, hipoxia, toxinas o arritmias cardíacas1, 2.
El síndrome convulsivo es una de las consultas neurológicas más frecuentes en la edad pediátrica3. Aproximadamente 120.000 niños tienen su primera crisis convulsiva en los Estados Unidos cada año; además, cerca del 10% de la población tendrá un episodio convulsivo en algún momento de su vida y del 2 al 4% tendrá recurrencia o presentará epilepsia1.
El primer paso en la valoración y manejo de una crisis convulsiva es determinar la causa, que hasta el 30% son provocadas, es decir, que son consecuencia de desencadenantes extra cerebrales, como hipertermia, hipoglucemia u otras alteraciones esporádicas y transitorias. Por el contrario, las convulsiones no provocadas son aquellas donde no hay un precipitante evidente que puede haber causado la crisis y que no está relacionada a fiebre, desórdenes hidroelectrolíticos, traumatismo de cráneo reciente, tumor del SNC, evento cerebro vascular, intoxicación exógena y trastornos metabólicos1, 4.
Las crisis convulsivas son uno de los trastornos neurológicos más comunes en la infancia. Se definen como una descarga neuronal paroxística anormal manifiesta clínicamente por trastornos motores, sensoriales, autonómicos y de comportamiento. Aproximadamente 120.000 niños tienen la primera crisis convulsiva en los Estados Unidos cada año. Uno por ciento de los niños tendrán una convulsión febril hasta los 14 años, y más del 50% de las personas epilépticas tienen la primera crisis convulsiva durante la infancia o en la adolescencia5.
Se puede definir como una alteración del cerebro caracterizada por la predisposición mantenida a generar crisis epilépticas (CE) y por las consecuencias neurobiológicas, cognitivas, psicológicas y sociales de esta alteración, y requiriéndose al menos la existencia de una CE. Es una de las enfermedades que más afectan a la calidad de vida del paciente6.
Es el trastorno neurológico de ocurrencia más común a nivel mundial. Las crisis epilépticas se definen como propagación anormal y descontrolada de actividad eléctrica en el cerebro, la cual puede tener uno o varios focos de origen7, 8.
Cerca de 10,5 millones de niños en el mundo tienen epilepsia y representan el 25% de la población que padece esta enfermedad. Las causas y la clínica son ampliamente variables. De los 3,5 millones de personas que anualmente desarrollan epilepsia, el 40% son menores de quince años y de ellos el 80% viven en países en vías de desarrollo.
En cuanto a la clasificación de las crisis epilépticas el 70% son del lóbulo temporal y el resto son extratemporales. Gracias a los avances terapéuticos; se ha logrado que el 70% de crisis sean controladas con medicamentos antiepilépticos. Sin embargo, un 30% de casos cuyas crisis no se controlan, se consideran como refractarias o fármaco-resistentes8.
Se llama síndrome epiléptico al conjunto de signos y síntomas que define una condición epiléptica única y el cual debe incluir más de un tipo de crisis. En todas sus manifestaciones clínicas, ocupa un lugar preponderante dentro de las enfermedades de interés en salud pública, por su frecuencia, morbilidad e importancia clínica. En las últimas décadas se ha logrado un vuelco radical en el pronóstico de quienes padecen Epilepsia. En la actualidad 70-80% deberían aspirar a una vida sana, gratificante, libre de crisis, llegando a ser útiles a sí mismos, y a sus conciudadanos. Un 10% adicional probablemente no consiga control total pero sí una reducción significativa de sus crisis. Entre el 5-10% de la población siguen padeciendo formas refractarias de muy difícil control, aunque también para ellos se trabaja en nuevas alternativas terapéuticas que en no pocas ocasiones han logrado mejorar su pronóstico3, 7, 9.
Se define como la persistencia de actividad epiléptica o la recurrencia de la misma sin recuperación de la situación basal durante más de 30 minutos. Algunos autores han introducido recientemente el término de estatus epiléptico precoz para aquella crisis de más de 5 minutos de duración, ya que es el tiempo a partir del cual se comienza con tratamiento farmacológico para yugular la crisis. La causa más frecuente de esta patología son las crisis febriles. Hasta en el 12% de los pacientes es la forma de presentación de la primera crisis. La mortalidad en niños se estima en el 2-5%, mucho menor que en adultos, dependiendo sobre todo de la causa del estatus y en menor medida de la duración del mismo3, 9.
La Clasificación Internacional de las Crisis Epilépticas (Comisión para la Clasificación y Terminología de la Liga Internacional contra la Epilepsia (ILAE-1981) divide las manifestaciones clínicas en crisis parciales (comienzan en un área hemisférica específica) y en crisis generalizadas (comienzan en los dos hemisferios simultáneamente)7, 22.
Cuando una crisis parcial no produce alteración de la conciencia se denomina crisis parcial simple. Si la conciencia está alterada se van a llamar crisis parciales complejas. Los síntomas o signos de las crisis parciales simples dependerán del área cortical involucrada en el foco epiléptico y se dividen en: motoras, sensitivas, autonómicas y psíquicas. Las crisis parciales simples sin síntomas motores son denominadas auras. Las descargas neuronales anormales focales pueden propagarse después de un tiempo desde su inicio y dar lugar a que la crisis parcial evolucione a una crisis parcial compleja o a una crisis generalizada, pasándose a llamar crisis parcial secundariamente generalizada.
Las crisis generalizadas pueden ser convulsivas y no convulsivas. Las convulsivas incluyen las tónicas, clónicas y tónico-clónicas. Las no convulsivas incluyen10:
Cuando cualquiera de las crisis anteriores produce caída al suelo, pueden causar traumatismos graves o una incapacidad muy importante. En ocasiones, el paciente puede notar síntomas prodrómicos sistémicos (malestar, nerviosismo, etc.) que marcan el inicio de una crisis generalizada y que no deben de ser considerados como crisis parciales que evolucionan a crisis generalizadas.
La epilepsia es una enfermedad que se caracteriza por crisis epilépticas recurrentes. Por lo tanto, una única crisis no constituye una epilepsia. Tampoco lo son las crisis reactivas a alteraciones transitorias del SNC que se resuelven espontáneamente o son tratadas satisfactoriamente8.
Las crisis epilépticas que aparecen como crisis reactivas son normalmente generalizadas, aunque pueden ser focales si ya el cerebro tiene una lesión3, 22.
En las crisis generalizadas hay una descarga simultánea, masiva, bilateral de actividad paroxística9. Las formas más conocidas son la ausencia (ruptura fugaz de contacto y detención de la actividad sin otros componentes o caída), crisis tónica (inconsciencia asociada a hipertonía generalizada como opistótonus), la clásica tónico-clónica, la crisis atónica (pérdida súbita y masiva del tono muscular con la consecuente caída, sin compromiso de conciencia y rápida recuperación) y la crisis mioclónica (contractura brusca, aislada y fugaz de algún segmento o grupo de segmentos)3, 7, 11, 12.
No es extraño que coexistan varios tipos de crisis. Cada uno debe ser registrado en sus características.

Imagen 1. Registro electroencefalográfico de una crisis generalizada
En la vida cotidiana de un individuo normal, la actividad de las distintas áreas de su cerebro "producen" toda la gama de expresiones humanas posibles, de acuerdo a una correspondencia entre estructura y función. Así por ejemplo, la actividad normal en el giro precentral del lóbulo frontal (homúnculo motor) "produce" movimiento en el hemicuerpo contralateral; la actividad en la cisura calcarina del lóbulo occipital y áreas aledañas generando reconocimiento visual entre otros. En la crisis epiléptica hay la misma actividad pero con carácter paroxístico, incontrolado, desordenado, involuntario y por lo tanto las mismas expresiones externas objetivables (movimiento clónico o automático de un segmento, alucinación visual)3, 12.
Consecuentemente, las posibles crisis focales son tantas como áreas funcionales existen en el cerebro (motoras, auditivas, visuales, somatosensoriales, del lenguaje, afectivas, autonómicas). Para ordenar y racionalizar esta variedad enorme de crisis está hoy universalmente aceptada la Clasificación Internacional de las Crisis Epilépticas10
Dentro de las crisis epilépticas podemos diferenciar las crisis parciales que se dividen en dos grupos: parciales simples, en las que no hay compromiso de conciencia, y parciales complejas en las que sí hay compromiso (variable) de la conciencia. Las parciales simples pueden ser motoras, sensoriales (auditivas, visuales...), autonómicas o psíquicas. Las parciales complejas pueden tener tan solo compromiso de la conciencia (ruptura de contacto únicamente) o también acompañarse de movimientos automáticos (chupeteo, frotarse las manos, caminar).En el 65% de los casos, la descarga en el cerebro no permanece restringida a su sitio de origen sino que se extiende a otras áreas y secundariamente se generaliza a todo el encéfalo; consecuentemente, las crisis parciales en la mayoría de los casos se extienden y aún se generalizan a tónico-clónicas3, 11.
Ello no las convierte en crisis generalizadas tónico-clónicas; se denominan entonces parciales que generalizan. Por ello precisar los primeros segundos de inicio de la crisis es muy importante.

Imagen 2. Registro electroencefalográfico de una crisis parcial.
La epilepsia es una causa importante de bruscas alteraciones en la conducta de un individuo, ya sea en su estado de alerta y conexión con el medio, en sus funciones motoras, sensitivas o sensoriales, como en la conducta social11, 12. Sin embargo, existe una larga serie de condiciones, la mayoría de ellas originadas también en el cerebro, que se manifiestan por síntomas episódicos, generalmente de aparición brusca y breve duración, que reconocen mecanismos distintos al fenómeno epiléptico. En conjunto, estos trastornos cerebrales paroxísticos y síntomas episódicos no epilépticos alcanzan una prevalencia del 10% en la población infantil. Al comparar esta cifra con el 1% de prevalencia de epilepsias en el mismo grupo etario, se comprende claramente el valor del diagnóstico diferencial por sus proyecciones pronósticas y terapéuticas3,9,13.
Los pediatras están familiarizados con los ataques que se inician en el curso de un llanto por frustración, dolor, temor o enojo. Después de uno o varios movimientos respiratorios durante el llanto, éste se interrumpe, el niño deja de respirar y tras unos segundos se pone cianótico y pierde el conocimiento. La pérdida de conciencia se asocia a hipotonía generalizada o puede presentarse hipertonía con opistótonos y posteriormente convulsiones. Esta secuencia puede darse en forma completa o incompleta y lógicamente la reacción de los padres, incluso de los pediatras, es distinta si al pequeño sólo se le corta el llanto, y adquiere una leve coloración cianótica, o si la apnea va seguida de pérdida de la conciencia y hasta de movimientos convulsivos9.
Los ES son prácticamente inconfundibles con crisis epilépticas para el especializado en trastornos convulsivos, pero es habitual la referencia de estos pacientes a servicios de neurología infantil por dudas en el diagnóstico9.
Por otra parte destacar otro tipo de ES, se trata del espasmo del sollozo de tipo pálido: después de un traumatismo leve o una situación de temor y sorpresa, el niño casi no alcanza a llorar y pierde el conocimiento con palidez e hipotonía generalizadas a veces seguida de breves sacudidas clónicas en las extremidades. Con frecuencia son provocados por traumatismo leves del cráneo en región occipital y por esta razón son es fundamental diferenciarlos de la pérdida de la conciencia por conmoción cerebral o de crisis epilépticas desencadenadas por traumatismos3, 9,13.
Es muy importante reconocer ambos tipos de ES, los de tipo cianótico y los de tipo pálido, pues si bien responden a mecanismos patogénicos distintos, pueden observarse en un mismo paciente y tienen las mismas connotaciones diagnósticas y pronósticas3, 9 ,13.
Los SIJ se observan en niños mayores, generalmente en edad escolar o en púberes. Los factores precipitantes son habitualmente situaciones de estrés emocional, temor o dolor. En ocasiones se puede detectar un estado de ansiedad, angustia, tensión emocional, sin que exista un factor desencadenante inmediato. También influye el cambio de decúbito pues algunos pacientes sólo presentan las crisis al pasar a la posición erecta y nunca se producen estando el sujeto acostado.
Los síntomas iniciales son mareo y visión borrosa. Además puede manifestarse sensación de frío y hormigueo en extremidades antes de la pérdida del conocimiento. Durante la crisis se detecta palidez, bradicardia, sudoración fría y pueden asociarse náuseas, vómitos, incluso incontinencia urinaria. Si la anoxia se prolonga más de 15 segundos se agrega un espasmo tónico generalizado con opistótonos o sacudidas mioclónicas. Los episodios más intensos pueden ir seguidos de sueño. En general los SIJ no duran más de 15 segundos y el hecho de tomar una posición horizontal hace que mejore el flujo circulatorio cerebral y no se produzcan las convulsiones por hipoxia.
El principal diagnóstico diferencial es la epilepsia, especialmente las crisis atónicas en que el enfermo se ve desplomado sobre sí mismo mientras pierde la conciencia, y ciertas formas de epilepsia temporal que confunden por sus componentes vegetativos y la presencia de desencadenantes emocionales. En cambio, en las crisis atónicas o mioclónicas-atónicas la caída es mucho más súbita y prácticamente no se detecta la pérdida de la conciencia10.
De todos modos el EEG normal es un elemento útil para apoyar la presunción clínica de crisis vagotónica. También está indicado el Tilt Test o prueba de inclinación cefálica brusca que provoca una bradicardia, confirmando la hipervagotonía refleja.
Aunque en la mayoría de los casos se trata de una condición benigna que no requiere tratamiento específico, estos ``desmayos´´ pueden ser la primera manifestación de una enfermedad cardiovascular. En estos casos es fundamental el diagnóstico, pues corre riesgo la vida del paciente. Los SIJ tienden a hacerse menos frecuentes y desaparecer hacia el final de la adolescencia, sin secuelas12, 13.
El reconocimiento de una crisis vertiginosa en la infancia no es fácil. Los niños mayores pueden precisar verbalmente cuáles son sus síntomas, pero en los más pequeños el vértigo puede manifestarse por una pérdida de equilibrio, una resistencia al movimiento, una tendencia a echarse al suelo o quedarse quietos, o bien encubiertos por una gran ansiedad.
Clínicamente se trata de niños de
Es habitual que se muestren atemorizados y durante la crisis se puede observar nistagmo, palidez, náuseas y ocasionalmente vómitos. Estos episodios duran por lo general 1 minuto y el niño conserva su lucidez sin mostrar obnubilación ni somnolencia. Al cesar el episodio, retoma su actividad normal9.
La frecuencia es variable pero en general se repiten con intervalos de varias semanas. El vértigo paroxístico benigno tiene una evolución espontánea hacia la curación clínica. En el curso de meses o pocos años las crisis disminuyen en intensidad y frecuencia, hasta que desaparecen totalmente en edad escolar.
Es importante destacar que se trata de niños sanos sin antecedentes significativos y que los episodios vertiginosos ocurren en pleno estado de salud 12, 13.
En lactantes vomitadores se producen, a veces, episodios asociados con la ingesta de alimentos caracterizados por sacudidas o contracciones tónicas de miembros superiores y tronco, con inclinación cefálica o sin ella, con apnea o sin ella, en ausencia de aspiración manifiesta. Se trata de pequeños entre 2 y 12 semanas de vida que tienen reflujo gastroesofágico asociado a hernia hiatal y el cuadro se conoce como síndrome de Sandifer. En realidad no es imprescindible que exista hernia hiatal, pues resulta suficiente para el diagnóstico la demostración de reflujo gastroesofágico por medio de la radiografía seriada, el esofagograma o los estudios de manometría esofágica. Ocasionalmente esta alteración puede provocar tortícolis en lugar de las crisis mencionadas. En todos los casos el tratamiento con alimentos espesos y posición semisentada ha permitido controlar los síntomas que semejaban un trastorno convulsivo3, 9, 12, 14.
Los trastornos de origen psíquico que simulan enfermedades orgánicas del sistema nervioso pueden tener expresión periférica como la parálisis, temblores y otros movimientos anormales, trastornos de la marcha, de la sensibilidad; o bien presentarse en forma de crisis con alteración del estado de la conciencia o cambios paroxísticos en la conducta. Sin entrar en clasificaciones psiquiátricas, cabe mencionar que las rabietas (expresiones físicas de enojo que tiene una clara motivación previa) son un trastorno de la conducta común en el niño pequeño, mientras que los ataques de pánico, crisis de rabia psicopática y los episodios histéricos son más frecuentes en adolescentes a partir de la pubertad9.
Algunas condiciones neurológicas no epilépticas y paroxísticas que se manifiestan como trastornos motores episódicos son3, 9, 11:
Suelen aparecen en las primeras semanas de vida. Las primeras mioclonías más sutiles de los primeros días pueden pasas inadvertidas ya que van en aumento hasta la tercera semana de vida. Aparecen de forma dominante durante el sueño inquieto. En la mayoría de los casos son evidentes en miembros superiores, pudiendo manifestarse también crisis axiales, en cara y en músculos abdominales. Las mioclonías pueden ser bilaterales o localizadas, rítmicas o arrítmicas, e incluso migratorias o multifocales. Desaparecen siempre al despertar y en ocasiones pueden ser estímulo-sensibles. Disminuyen en intensidad a partir del segundo mes de vida y habitualmente desaparecen antes de los seis meses.
Dado que se trata de un fenómeno transitorio que no requiere medicación, el reconocimiento de este síndrome y el diagnóstico diferencial con otros movimientos anormales en el período neonatal es crucial para evitar medicaciones y estudios innecesarios en estos bebés12, 13.
Suelen comenzar entre los 4 y 9 meses de edad y cursan con aparición de contracciones bruscas de la musculatura del cuello o de los miembros superiores predominantemente con flexión o rotación de la cabeza y extensión de los miembros superiores. Se caracterizan por maduración neuropsíquica y examen neurológico normal. Suele ocurrir varias crisis por día, en estado de vigilia, excepcionalmente en sueño y tienden a repetirse en salvas. El EEG es normal y no hay signos de deterioro psicomotor3, 9, 13, 14.
Se trata de una entidad definida de naturaleza no epiléptica. El cuadro clásico se caracteriza por la aparición temprana de reacciones de sobresaltos a diferentes estímulos. Estas respuestas parecen verdaderas mioclonías estímulo-sensibles, pero también pueden ser breves rigideces generalizadas con pérdida del control postural que lleva a la caída. Más aún en los lactantes se observa una hipertonía persistente durante el primer año de vida, con reflejos tendinosos vivos pero sin signos piramidales definidos. Estos niños luego tienen una marcha insegura y caen con frecuencia. Estas tensiones se incrementan por la tensión emocional y la fatiga. A menudo presentan además episodios durante el sueño, que semejan crisis generalizadas clónicas o mioclonías repetitivas14.
Los exámenes de laboratorio, el LCR, las prueba neurometabólicas, la tomografía computarizada cerebral y el EEG son normales.
Se trata entonces de una condición muy rara, que además se confunde en los primeros años con parálisis cerebral y epilepsia. La mejor medicación suele ser el clonazepán, que produce efectos sostenidos13.
Las convulsiones en niños asociadas a fiebre son conocidas desde la antigüedad. En los escritos hipocráticos se señalan los hechos fundamentales de ellas: fiebre, edad de aparición y susceptibilidad individual. “Las convulsiones ocurren en niños si hay fiebre aguda con mayor facilidad en su tercer año de vida. Los niños de mayor edad y los adultos no muestran esta propensión, a menos que precedan síntomas de mayor intensidad y peores”. Las convulsiones febriles constituyen actualmente uno de los más importantes problemas pediátricos en razón de su elevada prevalencia y de la controversia existente sobre su delimitación, pronóstico y tratamiento16.
Se define como convulsión asociada a enfermedad febril, en ausencia de infección del SNC, desequilibrio hidroelectrolítico o causa intracraneal definida.
Las convulsiones febriles (CF) son un problema común en la infancia, ya que hasta un 5% de los niños tiene el antecedente de al menos una CF. Constituyen la manifestación convulsiva más frecuente en los primeros años de vida. Pueden definirse como convulsiones desencadenadas por la fiebre, que no esté originada por una infección del sistema nervioso central, en niños de 6 meses a 5 años de edad sin anomalías neurológicas previas15, 16.
Las convulsiones febriles son un problema común en la infancia, dentro de la consulta neuropediátrica y un importante motivo en los servicios de Urgencia ya que hasta un 5% de los niños tiene el antecedente de al menos una convulsión febril15.
Son el problema más común. Afecta entre un 4-5% de los niños; ocupando el rango de edad que va desde los 6 meses a los 5 años de edad1. Su incidencia anual estimada es de 460 casos por cada 100.000 niños que consultan al Servicio de Urgencia, siendo levemente más frecuente en hombres que en mujeres (1,5:1)15.
Existen tres factores que interactúan en la producción de crisis convulsivas febriles15:
Las crisis febriles se clasifican en simples o complejas (tabla 1), siendo entre el 70% y 75% de las convulsiones febriles provocadas por crisis simples. A grandes rasgos, la crisis febril simple es aquella convulsión generalizada, de buen pronóstico y corta duración, que se dan en niños sin antecedentes previos de crisis y que no presenta otro episodio en las 24 horas posteriores al evento. Por otro lado, la crisis febril compleja posee una duración superior a 15 minutos, son de carácter focal, pudiendo afectar únicamente a un hemicuerpo y pueden repetirse en el mismo proceso febril dentro de las primeras 24 horas. Son estas últimas las que tienen mayor riesgo de complicación. Representan entre 9%-35% de las convulsiones febriles. Pueden presentarse como status epilepticus febril cuando la convulsión se prolonga por más de 30 minutos. Si bien esta presentación no es común (5%), constituye un 25% de los status epilepticus del niño2, 17
Tabla 1. Clasificación de las crisis febriles.
|
CRISIS FEBRIL SIMPLE |
CRISIS FEBRIL COMPLEJA |
|
< 15 minutos Generalizada Sin recidiva en las primeras 24 horas Sin historia previa |
> 15 minutos Focalizada Recidiva dentro de las primeras 24 horas. Daño neurológico previo, anormalidad del SNC y/o historia de crisis afebriles. |
Comienzan a menudo con un grito o llanto al cual le sigue la pérdida de conocimiento, el cual suele ser breve y se asocia a convulsiones que pueden ser de cualquier tipo, generalmente tónico-clónicas generalizadas y con menos frecuencia (4%) focales. La mayoría ocurre
Se hablará del diagnóstico y tratamiento en temas posteriores.
La EHI se produce como consecuencia de la deprivación de O2 al cerebro, bien por hipoxemia arterial o por isquemia cerebral, o por la concurrencia de ambas situaciones. El examen neurológico permite establecer la presencia o la ausencia de encefalopatía aguda. Se han diseñado una serie de esquemas de graduación que clasifican la profundidad de la EHI en distintos estadios. Estos esquemas reflejan el hecho de que cuanto mayor es el deterioro de la vigilia y de la capacidad para despertar, más grave es la encefalopatía26. La caracterización clínica de la gravedad de la EHI es un barómetro sensible de la gravedad de la agresión al SNC y tiene una importante utilidad pronóstica durante los primeros días de vida al correlacionarse estrechamente con la probabilidad de secuelas neurológicas. La EHI leve no conlleva ningún riesgo de mortalidad ni de minusvalía moderada o severa ulterior; aunque entre un 6% y un 24% presentan leves retrasos en el desarrollo psicomotor. En la EHI moderada, el riesgo de mortalidad neonatal es en torno al 3% y el de minusvalías moderadas o graves en los supervivientes muestra una amplia variabilidad; entre un 20% y un 45%. En la EHI severa, la mortalidad es muy elevada (50-75%) y prácticamente todos los supervivientes desarrollan secuelas neurológicas24,25.

Imagen 3. TC que muestra infartos secundarios a EHI.
La EHI está presente desde el nacimiento, no existiendo un periodo de tiempo libre de sintomatología clínica. El perfil neurológico evolutivo en el curso de los primeros días o semanas permite diferenciar la EHI perinatal de una encefalopatía de origen prenatal. Mientras la primera muestra un perfil dinámico o cambiante, la segunda muestra uno estable. Además, el curso temporal es de gran valor para establecer más certeramente el pronóstico. En general, en la EHI leve y moderada el cuadro clínico comienza a mejorar progresivamente después de las 72 horas de vida. Volpe ha descrito un síndrome neurológico postasfíctico grave, caracterizado por la presencia de estupor profundo o coma durante las primeras 12 horas de vida. Durante este tiempo generalmente el RN no presenta disfunción del tronco cerebral, está marcadamente hipotónico y presenta convulsiones sutiles y clónicas multifocales. Entre las 12 y las 24 horas de vida, parece mejorar el nivel de alerta, pero esta mejoría es más aparente que real, ya que no hay contacto con el medio y existe con frecuencia un estado epiléptico, siendo frecuentes las crisis tónicas. Entre las 24 y 72 horas de vida, parece agudizarse el deterioro de la capacidad para despertar y con frecuencia aparece disfunción del tronco encefálico y algunos RN presentan signos de hipertensión intracraneal. Es en este período cuando el neonato habitualmente fallece. Los que sobreviven experimentan una progresiva mejoría en la vigilia, el tono muscular cambia progresivamente de la hipotonía inicial a distonía o hipertonía extensora, y aparece una combinación de parálisis bulbar y seudobulbar que determina problemas en la alimentación. La progresión de la mejoría neurológica es variable y difícil de predecir, y se cree que aquellos que mejoran rápidamente pueden tener un mejor pronóstico24, 25, 26.
Las infecciones en el cerebro y la médula espinal pueden causar una inflamación peligrosa. Esta inflamación puede producir una amplia gama de síntomas, como fiebre, dolor de cabeza, o confusión y en casos extremos, puede causar daño cerebral, accidente cerebrovascular, convulsiones, o la muerte27.
La meningitis bacteriana es una enfermedad rara pero potencialmente mortal. Puede estar causada por varios tipos de bacterias que primero producen una infección de las vías respiratorias altas y luego viajan por el torrente sanguíneo al cerebro. La enfermedad también puede producirse cuando ciertas bacterias invaden directamente las meninges y puede bloquear los vasos sanguíneos cerebrales, causando un accidente cerebrovascular y daño cerebral permanente. Las bacterias más comunes causantes de meningitis son el neumococo y el meningococo27.
La meningitis neumocócica es la forma más común de meningitis y la forma más seria de meningitis bacteriana. Cada año se informan unos 6,000 casos de meningitis neumocócica en los Estados Unidos. La enfermedad está causada por la bacteria Streptococcus pneumoniae, que también causa la neumonía, sepsis, e infecciones sinusales y de los oídos. Los niños menores de 2 años y los adultos inmunodeprimidos se encuentran particularmente a riesgo. Las personas que tienen meningitis neumocócica a menudo sufren daño neurológico que varía desde la sordera al daño cerebral grave.
La meningitis meningocócica, causada por la bacteria Neisseria meningitidis, es común en los niños de 2 a 18 años. Anualmente en los Estados Unidos alrededor de 2,600 personas contraen esta enfermedad altamente contagiosa. Los grupos de alto riesgo comprenden a los bebés menores de 1 año, las personas con sistemas inmunitarios suprimidos, los viajeros a países extranjeros donde la enfermedad es endémica, y los estudiantes universitarios (en particular del primer año) que residen en dormitorios. Entre el 10 y 15 por ciento de los casos es mortal, con otro 10 a 15 por ciento que causa daño cerebral y otros efectos secundarios serios.
La meningitis viral o aséptica es la forma más común de meningitis en los Estados Unidos. Esta enfermedad típicamente leve que no es mortal está generalmente causada por enterovirus, virus comunes que entran al cuerpo por la boca y viajan al cerebro y los tejidos circundantes donde se multiplican. Los enterovirus están presentes en el moco, la saliva y las heces y puede transmitirse por contacto directo con una persona infectada o un objeto o superficie infectado. Otros virus que causan la meningitis son la varicela zoster, influenza, parotiditis, VIH, y herpes simple. La encefalitis puede estar causada por una infección bacteriana y, más frecuentemente, por infecciones virales. Anualmente se informan varios miles de casos de encefalitis, pero realmente pueden producirse muchos más ya que los síntomas pueden ser leves a no existentes en la mayoría de los pacientes.

Imagen 4. Resonancia magnética de encefalitis por virus herpes simple.
La mayoría de los casos de encefalitis en los Estados Unidos está causada por enterovirus, virus del herpes simple tipos 1 y 2, el mordisco de un animal rabioso (virus de la rabia), o arbovirus, que se transmiten de animales infectados a humanos por la picadura de una garrapata infectada, un mosquito, u otro insecto que succiona sangre. La enfermedad de Lyme, una infección bacteriana diseminada por la picadura de la garrapata, puede causar encefalitis27.
La existencia de asociación entre convulsiones afebriles y gastroenteritis aguda (GEA) viral leve era una entidad poco conocida fuera del continente asiático. Actualmente están apareciendo artículos sobre esta patología realizados en otros continentes como Europa y América. Este tipo de convulsiones en niños previamente sanos sin alteraciones neurológicas, no asociadas a fiebre, deshidratación ni desequilibrio hidroelectrolítico importante en el contexto de una gastroenteritis aguda viral, se caracterizan por tener un carácter benigno con buen pronóstico sin precisar tratamientos específicos prologados, por lo cual es importante conocer su existencia e identificarlas.
El rotavirus es el principal causante de GEAs virales en lactantes y además es el principal agente responsable de esta entidad. Se conoce que las infecciones por rotavirus, aunque es raro, pueden asociarse con manifestaciones neurológicas: encefalitis-cerebelitis o convulsiones afebriles. Existe, además, mayor riesgo de convulsiones si la GEA es debida al rotavirus, aunque otros virus como los enterovirus, astrovirus, calicivirus, también se han identificado como asociados a convulsiones afebriles benignas. Las GEAs por rotavirus tienen un mayor riesgo de encefalopatía si las comparamos con las GEAs bacterianas. La fisiopatología por la que se producen este tipo de convulsiones es desconocida28, 29.
Numerosos errores congénitos del metabolismo (ECM) pueden presentar crisis epilépticas en los primeros años de vida (aunque rara vez las crisis son la manifestación más importante). De todas maneras es preciso recordar que las causas que provocan crisis en los recién nacidos no suelen ser de origen metabólico: hipoxia, hemorragia cerebral, infección, patología cardiopulmonar, malformaciones cerebrales. El reconocer a una ECM como causa de epilepsia es fundamental no sólo para un correcto consejo pronóstico y genético, sino porque en algunos casos es posible un tratamiento específico curativo30.
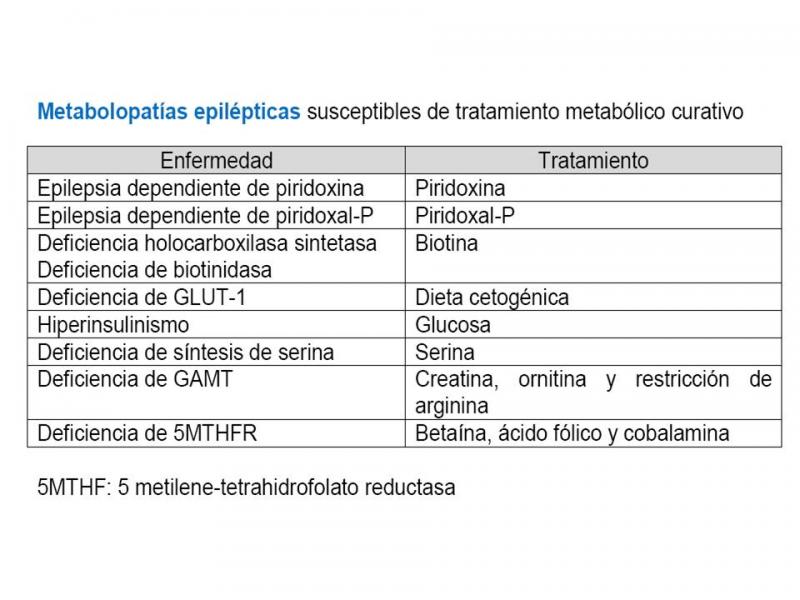
Tabla 2. Metabolopatías susceptibles de tratamiento.
Por tanto, ante un recién nacido con crisis epilépticas de causa desconocida hay que valorar iniciar tratamiento con30:
Las malformaciones congénitas del sistema nervioso central están originadas por un insulto al embrión durante el embarazo. Su etiología es multi-factorial: se consideran factores genéticos, hipóxicos y sustancias inflamatorias nocivas.
Ocurren en aproximadamente 0.1-0.9% de los nacimientos. Las malformaciones de la columna vertebral y médula es-pinal (espina bífida, mielocele, mielomeningocele y ra-quisquisis) en general aparecen una por cada 1,000-2,000 nacimientos. En China, en 4,628 pacientes con malformaciones del SNC, se encontró una prevalencia de defectos del tubo neural al nacimiento de 27.37 y 37.22 por 10,000, predominando en mujeres (35.68).
Dentro de las malformaciones congénitas, tienen especial relevancia las malformaciones arteriovenosas, ya que pueden ser causantes de convulsiones. La mayoría de la gente que padece de malformaciones arteriovenosas presenta muy pocos síntomas de importancia y las malformaciones tienden a ser descubiertas sólo por casualidad, usualmente durante una autopsia o en tratamientos por causas no relacionadas. Sin embargo, en aproximadamente el 12 por ciento de la población afectada (cerca de 36 mil de los 300 mil estadounidenses que se estima que padecen de MAV) estas anomalías, llamadas también lesiones, causan síntomas cuyo grado de severidad varía considerablemente. En un pequeño número de individuos en este grupo, los síntomas son lo suficientemente graves como para causar debilitamiento o inclusive pueden provocar la muerte. Anualmente, aproximadamente el 1 por ciento de las personas que padecen de MAV mueren como consecuencia directa de estas lesiones31.

Imagen 5. Representación de una malformación arteriovenosa cerebral.
Los síntomas más generalizados de las MAV incluyen convulsiones y dolores de cabeza, pero no se ha identificado un patrón específico de estos síntomas. Las convulsiones pueden ser parciales o totales, pueden ocasionar una pérdida de control en el movimiento o un cambio en el nivel de conciencia de la persona. Los dolores de cabeza pueden variar significativamente en frecuencia, duración e intensidad, llegando a veces a ser tan graves como las migrañas. En ciertos casos, un dolor de cabeza que afecta de forma constante un solo lado de la cabeza puede ser atribuido directamente a la ubicación de una malformación arteriovenosa. Con mayor frecuencia, sin embargo, la ubicación del dolor no tiene relación directa con la lesión y puede abarcar la mayor parte de la cabeza31.
Las intoxicaciones constituyen un problema frecuente y muchas veces grave. En la infancia suponen el 0,3% de las asistencias pediátricas. Son una causa importante de convulsiones32, 33.
Las intoxicaciones accidentales, intencionales y las sobredosis de drogas constituyen un grupo importante de enfermedades con alta morbilidad, mortalidad y costos en salud. Se estima que existen anualmente en Estados Unidos entre 25 millones de intoxicaciones y sobredosis de drogas33.
Aunque al principio el paciente intoxicado no luzca enfermo, los pacientes intoxicados deben ser tratados como si tuvieran una enfermedad potencialmente mortal, hasta que el diagnóstico específico y su evolución demuestren lo contrario.
Debemos sospechar intoxicación en un paciente que se presenta con32, 33:
Es importante destacar que el 4% de las admisiones hospitalarias por toxicología requieren hospitalización. En cuanto a las exposiciones a tóxicos, más del 5% de pacientes requieren ingreso a una Unidad de Cuidado Intensivo.
- Intoxicaciones por bebidas energéticas.
En la adolescencia, las intoxicaciones más frecuentes suelen ser de etiologia accidental y en el marco del ocio. En este rango de edad, es importante resaltar el consumo de las llamadas bebidas energéticas, con alto contenido en cafeína, extracto de guarana, taurina, ginseng y otras sustancias estimulantes que, de forma abusiva son consumidas por un alto porcentaje de jóvenes entre los 13 y 15 años. Dicho consumo puede llevar a causar riesgo de sobredosis de cafeína en niños y adolescentes.
Solas o combinadas con otras drogas, pueden provocar cuadros de psicosis aguda, manía o cuadros de agitación psicomotriz intensa y arritmias cardiacas severas. Además de la cafeína, otras sustancias incluidas en estas bebidas pueden interaccionar con algunos fármacos disminuyendo o potenciando su efecto.
- Intoxicaciones medicamentosas.
Cabe destacar las producidas por la ingesta de paracetamol, fármaco muy utilizado en la infancia fuera del ámbito clínico y al alcance de la población. En un reciente estudio, en 44 niños hospitalizados se detectó un 68% de intoxicaciones intencionales, un 22,7% accidentales y un 4% por sobredosificación, por lo que debe alertarse del acceso usual e ilimitado de los pacientes al paracetamol. Otros medicamentos como los antieméticos, muy comunes en Pediatría, son usados con frecuencia en la infancia. La ciclizina a dosis altas puede provocar una intoxicación que da origen a convulsiones. Los facultativos deben conocer e informar sobre la limitación de estos productos y la dosis utilizada cuando no hay prescripción médica especifica32, 33.
- Intoxicación por drogas de abuso
En Pediatria, las intoxicaciones por drogas de abuso durante los primeros años pueden derivarse de la exposición a las mismas durante el embarazo (etapa prenatal), a través de la lactancia materna o bien por exposición (etapa posnatal). Por otro lado, en la adolescencia, el origen suele ser un consumo directo o abuso con finalidad recreativa. Cuando los padres son consumidores de drogas, la exposición durante el periodo prenatal puede extenderse a los primeros años de la vida y afectar al menor por vía inhalatoria, ingestión accidental o contaminación del entorno material en muebles o enseres, dando lugar a intoxicaciones agudas o crónicas32, 33.
El trauma craneoencefálico es aquel en el cual el episodio traumático genera alteración funcional o estructural del encéfalo, expresado con una puntuación inicial en la Escala de Coma de Glasgow menor de 13 puntos34,35.
El trauma pediátrico es un problema de salud pública y la principal causa de morbi-mortalidad en niños. Presenta una incidencia elevada y continúa siendo una de las principales causas de muerte y discapacidades permanentes en niños34,35,36.

Imagen 6. Mecanismo de trauma por impacto.
En la actualidad se reconoce el TCE como una enfermedad, una lesión traumática provocada por una causa externa prevenible causada por el daño al organismo debido a la brusca exposición de una concentración de energía que supera un margen de tolerancia del niño y a politraumatismo como el daño corporal sufrido a consecuencia del intercambio de energía que se produce en un incidente y que afecta a uno o varios órganos o sistemas con la magnitud suficiente como para poner en peligro la vida del paciente.
Las causas del TCE varían según la edad, en niños menores de 2 años se deben a caídas y maltratos, entre 2 y 10 años son por accidentes de tránsito, caídas y accidentes en bicicleta, y en niños mayores de 10 años debido a deportes, accidentes de tránsito y accidentes en bicicleta, siendo los accidentes de tránsito la causa del traumatismo craneal grave más frecuente en todos los grupos etarios36.
Cabe destacar que los niños presentan diferencias anatómicas y fisiológicas respecto a los adultos, por lo que requieren un cuidado especial durante la atención del trauma avanzado.
Los estudios de imagen son de gran utilidad para identificar las lesiones en los infantes. La radiografía simple de cráneo es capaz de detectar del 94 al 99% de las fracturas lineales deprimidas, mostrando superioridad frente a la TC cuya sensibilidad varía del 47 al 94%; sin embargo, posee utilidad limitada para detectar lesiones intracraneales, ya que se ha demostrado que no existe fractura evidente en la radiografía simple en la mitad de los pacientes que presentan lesiones por TC.7 Además, la tomografía de cráneo ha mostrado gran utilidad en las lesiones que precisan de atención quirúrgica inmediata y brinda información pronóstica del paciente, lo que convierte a este estudio en la técnica de elección en el diagnóstico de las lesiones asociadas a TCE.8,9 Los pacientes con TCE de bajo riesgo no precisan estudio con TC. Se recomienda realizar tomografía a los niños que han presentado un traumatismo si presentan un Glasgow menor a 1334, 35, 36.
El tumor cerebral es una masa formada por el crecimiento de células anormales o la proliferación incontrolada de dichas células en el cerebro. Los cánceres primarios involucran a cualquier masa que se origina en esta parte del sistema nervioso central (SNC) y no a aquella que se disemine hasta esta zona desde otra parte del cuerpo37.
Los tumores del SNC constituyen la segunda causa de muerte en los menores de 15 años. Representan 15-20 % de todas las neoplasias de la infancia y la adolescencia; su incidencia varía entre 2-19 por cada 100 000 personas. Predominan en el sexo masculino en una relación de 1,2:1 con respecto al femenino37,39.
Los tumores primarios malignos del sistema nervioso central (SNC) son los tumores sólidos más comunes de la infancia y después de las leucemias, ocupan el segundo lugar en frecuencia. El tratamiento de los tumores cerebrales es complejo por la diversidad histológica de las lesiones y la tendencia de la mayoría de ellos a diseminar en el neuroeje precozmente en el curso de la enfermedad. Además, pueden producirse secuelas importantes secundarias a las intervenciones terapéuticas39.
Las crisis convulsivas son el primer síntoma del 6-10% de los tumores cerebrales infantiles, y aparecen a lo largo de la evolución en un 10-15% adicional. Su aparición depende de la localización tumoral (50% de los tumores hemisféricos producen convulsiones), de la estirpe celular (gangliogliomas y astrocitomas especialmente), del grado de malignidad (en menores de 10 años, 28% de los gliomas de bajo grado debutan con epilepsia, y 12% de los alto grado), y de la edad (el debut epiléptico de los tumores es menos probable en la infancia que en la edad adulta). Los pacientes tumorales que debutan con convulsiones tienen una exploración neurológica inicialmente normal en el 75% de los casos, y refieren alteraciones del comportamiento el 50%. Por otra parte, el 1-5% de la población epiléptica presenta una etiología tumoral, aumentando el porcentaje con la mayor utilización de estudios neuroradiológicos. El origen tumoral es más frecuente en las crisis parciales y en sujetos de menor edad: 17% de los menores de 4 años con epilepsia parcial. La larga evolución de la epilepsia disminuye el riesgo pero no lo suprime: OR 9,4 para las epilepsias con menos de un año de evolución y un 4,7 para aquellas con más de 10 años. Las convulsiones febriles no suponen riesgo alguno. Las crisis epilépticas pueden aparecer a cualquier edad, desde los primeros días de vida. Es importante la práctica de una RNM en todo niño con epilepsia parcial que no corresponda a los bien conocidos síndromes idiopáticos, y cuya etiología no haya sido firmemente establecida. En tales casos, la normalidad EEG o buena respuesta al tratamiento, no son garantía de ausencia tumoral.
En los niños sometidos a cirugía por epilepsia intratable, el porcentaje de tumores es del 12%, aumentando considerablemente la frecuencia si la serie quirúrgica se circunscribe al lóbulo temporal. Los pacientes con epilepsia intratable en los que la cirugía de la epilepsia demostró un origen tumoral tenían una exploración normal previa en el 80-97%. Las crisis son del tipo parcial complejo en la mitad de los casos, y en casi la tercera parte pueden asociarse diferentes tipos de crisis. Varios estudios han descartado la posibilidad oncogénica de los fármacos antiepilépticos. Toda la epilepsia de difícil control debe ser estudiada con RNM. Una TAC previa normal o con hipodensidades aparentemente residuales no supone garantía alguna en estos casos37,38.
La tomografía axial computarizada (TAC) permite la valoración del tumor proporcionando la información necesaria sobre la presencia o ausencia de este, tales como tamaño, forma y densidad tumoral, localización, manifestación después de administrar el contraste, calcificaciones, zonas de necrosis y quistes, edema peritumoral, desplazamientos y herniaciones cerebrales, afectación de estructuras óseas, presencia de hidrocefalia, así como hemorragia tumoral. Igualmente es imprescindible en el periodo posoperatorio para la detección de complicaciones entre las cuales figuran: neumocefalia, hemorragia, hidrocefalia y el seguimiento de recaídas37,38,39.

Imagen 7. Tumor cerebral primario que afecta el córtex motor, provocando convulsiones.
Ahora bien, la resonancia magnética nuclear supone un mejoramiento diagnóstico con respecto al TAC, puesto que proporciona una mejor definición tumoral y visibiliza neoplasias que antes estaban ocultas por las estructuras óseas de la base del cráneo, entre ellas los tumores del tronco y del ángulo pontocerebeloso. También proporciona imágenes en los 3 planos del espacio, lo que permite una mejor planificación para la cirugía. Está indicada para el seguimiento de los procesos expansivos, la detección de recidivas y el diagnóstico diferencial de complicaciones, tales como radionecrosis y diseminación tumoral. La angioresonancia brinda información sobre la naturaleza y la vascularización de los tumores, así como permite detectar la invasión de estructuras vasculares por la neoplasia. La resonancia funcional posibilita valorar las zonas lesionadas por el tumor37,38,39.
El estatus epiléptico (SE) es uno de los mayores problemas en la salud pública de todo el mundo. Constituye una emergencia médica frecuente, con costes elevados para la salud y elevada morbimortalidad. En la población pediátrica se hace más importante por las diferencias en cuanto al enfoque diagnóstico17.
Representa aproximadamente el 3,5% de los ingresos a las Unidades de Cuidados Intensivos (UCI) y hasta el 15% de los pacientes internados en Servicios de Neurología. La tasa de incidencia anual del SE basada en diferentes estudios realizados en Europa y Estados Unidos varía de
La mortalidad a largo plazo aumenta en los casos de etiología sintomática aguda con respecto de los casos de SE idiopáticos o criptogénicos, y el riesgo de déficits neurológicos después de sufrir un SE es más alto en los casos de SE de etiología aguda sintomática o causados por patología neurológica progresiva, comparado con el SE de causa idiopática/criptogénica o con los casos de SE febril6.
El estado epiléptico (EE) fue definido por la Liga Internacional contra la Epilepsia (ILAE) como una crisis que no muestra síntomas clínicos de detención después de una duración mayor a la que abarca la gran mayoría de crisis de ese tipo en la mayoría de los pacientes, o crisis recurrentes sin reanudación de la función base del sistema nervioso central (SNC) 19.
La ILAE en 1981 define Estatus Epiléptico como: “convulsión prolongada por 30 min, o convulsiones recurrentes sin recuperación de conciencia entre ellas que duran más de 30 min”.
La Organización Mundial de la Salud (OMS), aludiendo mecanismos fisiopatológicos, lo define como crisis epilépticas suficientemente prolongadas o repetidas como para provocar una condición fija y duradera. Lowenstein (1999) propone una definición operacional, aplicable principalmente a estatus epiléptico convulsivo generalizado, como convulsiones continuas que superan los 5 minutos, considerando que si persisten más de 5 minutos es probable que continúen más de 30 minutos, siendo estas últimas más difíciles de tratar, y recalcando la importancia del manejo precoz, para evitar progresión hacia estadios refractarios y sus secuelas19, 20.
Se caracteriza por crisis epiléptica que dura más de 30 minutos o como la falta de recuperación del estado de la conciencia entre varias crisis.
Inicialmente el estatus podría ser un tiempo que oscila entre 25 y 30 minutos; sin embargo, publicaciones y estudios recientes demuestran que el daño neuronal y sistémico puede ocurrir más temprano. Estudios han mostrado cómo la duración de una crisis epiléptica focal en niños es en promedio de 97 segundos y que si la crisis pasa de 5 minutos es poco probable que ceda espontáneamente, lo cual en la nos obliga a tomar decisiones más tempranas y a aceptar que si un paciente cursa con crisis que dure más de 5 minutos debe manejarse de forma agresiva para evitar complicaciones, que aumentan en forma proporcional al tiempo transcurrido hasta el control de las crisis.
El estado de mal epiléptico refractario se define como crisis con duración mayor de 1 hora o crisis recurrentes a razón de dos o más por hora sin recuperación del estado de conciencia a pesar del tratamiento con medicación antiepiléptica convencional. Sin embargo, desde el punto de vista clínico es preferible considerar refractario el estado epiléptico en el cual el paciente no responde a la terapia de primera línea6.
En nuestro país no existen datos concretos acerca de la frecuencia o morbimortalidad del status epiléptico. En el mundo, Estados Unidos (EU), Inglaterra, Suiza y Alemania lideran las estadísticas en cuanto a publicaciones de estado epiléptico prospectivo, esta condición en la población general es de 41 por 100.000 habitantes/año. Se estima que la incidencia en la población pediátrica está entre 18-41 por 100.000 habitantes/año. En los diferentes grupos pediátricos se ha encontrado que las causas varían: el estado epiléptico febril se presenta con mayor frecuencia en niños entre 1 y 2 años, mientras que entre los de 5 y 10 años la etiología predominante es el sintomático remoto, más aún en las poblaciones de bajos recursos. La mortalidad del estatus epiléptico en la población pediátrica está entre el 1 y el 8%6, 18.
El factor de riesgo más importante es tener diagnóstico previo de epilepsia; sin embargo, la edad temprana, la predisposición genética y las lesiones cerebrales adquiridas son otros importantes factores de riesgo10.
Las causas del estado epiléptico son muchas y variadas. Se puede clasificar en3,9:
El SE puede clasificar en6, 18: (tabla 2)
La mortalidad a corto plazo de EEC se reporta en 2,7-5,2%, hasta 8% en UCI, directamente relacionada con la causa, siendo hasta 2% por el SE propiamente dicho, y entre 12,5 y 16% en pacientes con causa sintomática aguda. En este grupo la cifra se eleva hasta 22,5% en menores de 2 años.
Tabla 3. Clasificación del estatus epiléptico
|
FOCAL |
Simple Complejo |
|
|
ESTATUS EPILÉPTICO CONVULSIVO |
GENERALIZADO |
Tónico clónico Mioclónico Tónico Clónico |
|
FOCAL |
Simple Complejo |
|
|
ESTATUS EPILÉPTICO NO CONVULSIVO |
GENERALIZADO |
Ausencias típicas Ausencia atípicas (sd de Lennox Gastaut) |
1. SE precoz: 5 minutos, momento de iniciar las medidas terapéuticas que se detallarán.
2. SE establecido: 30 minutos.
3. SE refractario (SE-R): no responde a tratamiento de 1° o 2° línea, entre 60 y 120 minutos, requiere anestesia general.
4. SE súper-refractario (SE-SR) si, tras 24 h de anestésicos, el SE continua o recurre, incluyendo recaídas durante la reducción o retiro de anestésicos9.
El estatus epiléptico refractario (EER) es aquel en el cual, las crisis persisten más de
Los pasos terapéuticos en el EER son6, 9:

En función de la sospecha diagnóstica:
Además de estas pruebas dirigidas a identificar una causa orgánica, en todo niño con una primera crisis afebril está indicada la realización no urgente de EEG y valoración por neuropediatría.
Exigen ingreso hospitalario y un estudio más amplio para descartar enfermedad orgánica. A veces pueden ser útiles y difíciles de reconocer. Se debe realizar analítica completa (HRF, monograma con calcio y fósforo, gasometría, láctico y amonio), ecografía transfontanelar (resonancia magnética nuclear durante el ingreso) y examen de líquido cefalorraquídeo3
Constituyen el fármaco de primera elección en el tratamiento de una convulsión, su penetración al cerebro es inmediata; poseen diversos sitios de acción pre sináptica, pos sináptica y no sináptica que aumentan la inhibición GABAérgica; el efecto final es una rápida inhibición de las descargas epileptogénicas y de la actividad clínica epiléptica. Dentro de las benzodiazepinas, las más usadas en clínica son las siguientes3, 9, 23:
Indicada como segundo escalón, tanto en crisis que no ceden tras dos dosis de benzodiazepinas (para frenarla), como en aquellas que ceden tras dicha segunda dosis (para intentar prevenir recurrencias). Alcanza su pico a los 15 minutos. No es útil en las crisis generalizadas. Su infusión requiere un manejo lento debido a su potencial de riesgo de desencadenar arritmias cardiacas. Su dosis de impregnación es de 15-20 mg/kg y debe diluirse en 20-100ml de SSN 0,9%. Presenta riesgo de hipotensión22, 23.
Tiene un amplio espectro de actividad anticonvulsiva, principalmente por sus efectos GABA A al potenciar sus efectos inhibitorios al aumentar el tiempo de apertura del canal del cloro sin cambiar su frecuencia de despolarización. Indicado en crisis focales y generalizadas, sin embargo presenta menor eficacia que las benzodiazepinas. La dosis es de 10-20 mg/kg. También está indicado ante una convulsión en el periodo neonatal (primera línea en neonatos), en la que se ha de corregir los trastornos electrolíticos y metabólicos. Se presentan como efectos adversos la sedación, hipotensión, rash cutáneo y depresión respiratoria22, 23.
Indicado cuando fracasan los fármacos anteriores. Diferentes estudios reportan una efectividad del 78-100% en estatus refractarios sobre los fármacos de primera línea. Causa raramente hepatotoxicidad, pero este riesgo aumenta en menores de 2 años, pacientes polimedicados, hepatópatas y metabolópatas conocidos. La dosis es de 60 mg/kg/día, comenzando por una dosis de 15-45 mg/kg a una velocidad de infusión de 1,5-3 mg/kg-min. En caso de no contar con la presentación endovenosa, puede utilizarse también el jarabe oral en las mismas dosis para aplicación intrarrectal22, 23.
Antiepiléptico de nueva generación recomendado como alternativa al ácido valproico en el estatus refractario. Tiene como ventajas la ausencia de efectos adversos que sí presentan los fármacos anteriores. En particular, es efectivo en combinación con las benzodiazepinas. Dado que su metabolismo no se encuentra asociado al hígado, podría ser el medicamento de elección en episodios asociados a trastornos metabólicos como la porfiria 3, 21, 22.





